Aspectos generales de la enfermedad
La MPS I en un trastorno genético progresivo, debilitante e infradiagnosticado
La MPS I es una enfermedad autosómica recesiva compleja que causa daños progresivos e irreversibles en prácticamente todos los sistemas y tejidos del organismo.
La MPS I forma parte de un grupo de siete trastornos genéticos estrechamente relacionados denominados en conjunto mucopolisacaridosis, y está causada por la deficiencia de la enzima lisosómica α-L-iduronidasa. Este defecto provoca una acumulación progresiva de glucosaminoglucanos (GAG) en las células, lo que causa complicaciones generalizadas en múltiples sistemas1,2.
En ausencia de tratamiento, las personas afectadas experimentan un deterioro potencialmente mortal de los sistemas musculoesquelético y cardiorrespiratorio y, en caso de afectación grave, neurodegeneración progresiva con deterioro cognitivo2-4. Debido a la presentación clínica variable y a la rareza de la enfermedad, es habitual una demora de varios años en el diagnóstico y tratamiento, lo que disminuye considerablemente la calidad de vida de las personas afectadas y puede empeorar el desenlace clínico2-4. Es fundamental establecer el diagnóstico y el tratamiento de inmediato para preservar la función de los órganos y evitar algunas consecuencias irreversibles2-5.
Fisiopatología
La MPS I es uno de los más de cincuenta trastornos hereditarios raros que reciben el nombre de enfermedades de depósito lisosomal. Cada una de estas enfermedades está causada por un defecto genético congénito que provoca la deficiencia de una o varias enzimas lisosómicas concretas. La edad de inicio, los órganos y sistemas afectados, así como la intensidad de estos trastornos varían notablemente entre sí, pero todos son progresivos.
La MPS I es una enfermedad hereditaria autosómica recesiva que cursa con manifestaciones anatomopatológicas en la mayoría de los sistemas y tejidos del organismo1. La enfermedad está causada por un defecto en el gen que codifica la enzima lisosómica α-L-iduronidasa que hace que las personas afectadas sean incapaces de producir la enzima o la produzcan en cantidades bajas. La consiguiente deficiencia de α-L-iduronidasa altera la capacidad de las células para degradar los glucosaminoglucanos (GAG) dermatán y heparán sulfato, lo que provoca la acumulación constante de GAG en las células1. Esto desencadena una lenta cascada de procesos patológicos específicos en los órganos y la pérdida de su función6. A medida que progresa la enfermedad, la mayor parte del daño celular y tisular se vuelve irreversible.
Al nacer, los bebés afectados parecen inicialmente normales desde el punto de vista clínico, pero con el tiempo, la acumulación progresiva de GAG provoca un deterioro de las funciones motriz, respiratoria y cardíaca y un aumento del tamaño de algunos órganos, como el hígado y el bazo1,7. Aunque presenten un grado semejante de deficiencia enzimática, los pacientes con MPS I pueden experimentar una amplia variedad de síntomas de grado muy variable1,2.
Progresión y pronóstico
Debido a la acumulación crónica y progresiva de glucosaminoglucanos en los lisosomas de todas las células del organismo, los pacientes afectados por la MPS I presentan una disfunción multiorgánica que conlleva una morbilidad considerable y una mortalidad prematura en los más gravemente afectados. Al igual que la mayoría de las demás enfermedades metabólicas, la MPS I es muy heterogénea, de modo que la edad de comienzo, los órganos y sistemas afectados, la gravedad y la velocidad de progresión de la enfermedad varían considerablemente.4
En los lactantes y niños pequeños gravemente afectados, los síntomas aparecen poco después del nacimiento y progresan rápidamente.4 Sin tratamiento, aproximadamente el 75% de estos niños fallecen antes de cumplir 10 años,8 habitualmente por enfermedad obstructiva de las vías respiratorias, infecciones respiratorias y complicaciones cardíacas.1
En niños y adultos con formas atenuadas de MPS I, la evolución de la enfermedad es mucho más variable. Las manifestaciones aparecen al principio de la infancia, progresan más despacio y son menos evidentes que en los pacientes gravemente afectados. Los pacientes con MPS I atenuada pueden vivir hasta la edad adulta, pero con una morbilidad importante y una disminución en la esperanza de vida.1
Reconocer las manifestaciones tempranas más frecuentes de los diferentes fenotipos de la MPS I es clave para su diagnóstico
Los pacientes con MPS I presentan signos y síntomas muy diversos4
|
|
Forma grave |
Forma atenuada |
Forma atenuada |
|
Manifestaciones frecuentes |
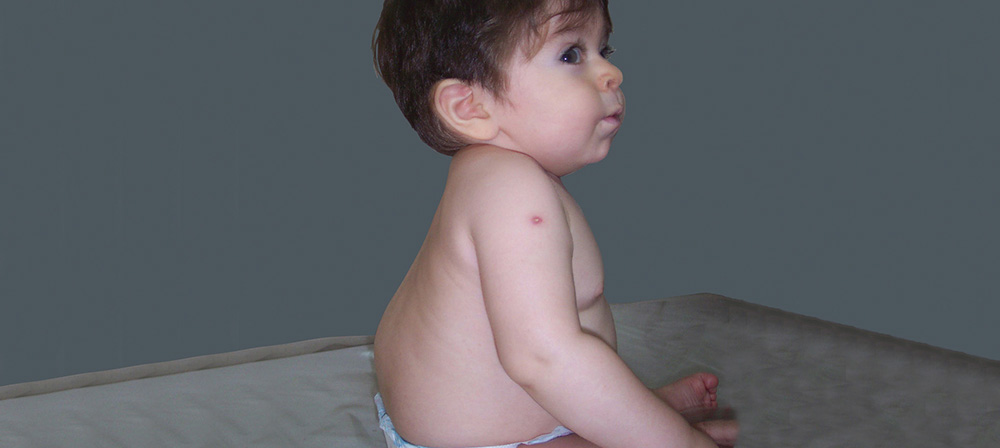
Cifosis/deformidad de giba
|
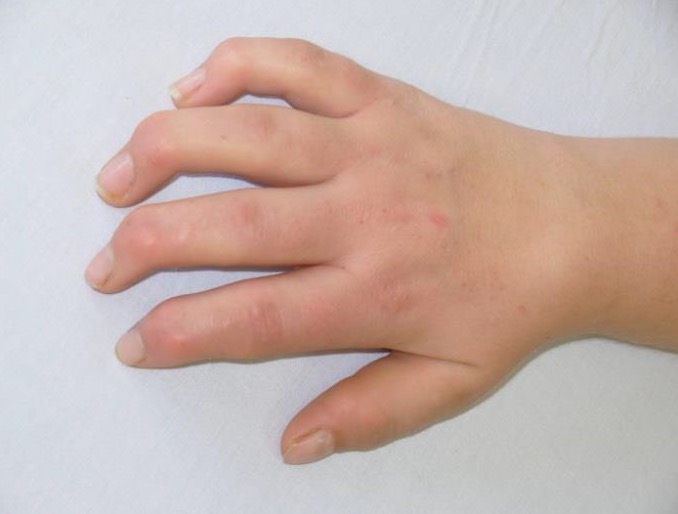
Contracturas articulares
|
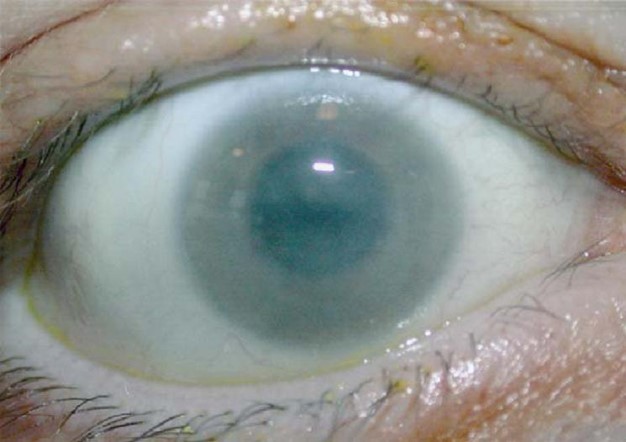
Opacidad corneal
|
| Otras manifestaciones |
Hernias, opacidad corneal, otras deformidades óseas, rasgos faciales toscos, hepatomegalia y alteraciones del sueño |
Alteraciones de las válvulas cardíacas, hepatomegalia, síndrome del túnel carpiano y hernias |
Alteraciones de las válvulas cardíacas, hepatomegalia, síndrome del túnel carpiano y hernias |
Fotografía por cortesía del Dr. Guelbert
Las formas atenuadas de la MPS I afectan a distintos órganos y sistemas. La edad de aparición de los síntomas es muy variable.

La acumulación de GAG en las células comienza al principio de la vida y progresa hasta provocar complicaciones debilitantes.

|
SÍNTOMAS INICIALES |
COMPLICACIONES TARDÍAS |
|
Hernias |
Enfermedad obstructiva de las vías respiratorias |
|
Rigidez/dolor articular |
Pérdida de movilidad |
|
|
Retraso del crecimiento |
|
Opacidad corneal |
Pérdida de visión, ceguera |
|
Soplo cardíaco |
Compresión medular |
|
Infecciones ORL |
Sordera |
|
Facies tosca |
Valvulopatía cardíaca grave |
|
Apneas del sueño |
|
|
Síndrome del túnel carpiano |
|
|
Astenia |
|
|
Retraso desarrollo (solo en la forma grave) |
Presentación clínica
La MPS I afecta a múltiples órganos y sistemas, con una gran variación fenotípica entre los distintos individuos. Hay un amplio espectro de intensidad de la enfermedad, y la edad de aparición de los síntomas oscila entre la infancia y la adolescencia. Históricamente, la MPS I se ha dividido en tres fenotipos definidos subjetivamente, que van desde el fenotipo grave y rápidamente progresivo (síndrome de Hurler) hasta los fenotipos atenuados de progresión más lenta (síndromes de Hurler-Scheie y Scheie).1 Sin embargo, hay un solapamiento considerable de síntomas entre los fenotipos, por lo que cada persona afectada presentará una evolución clínica diferente y un conjunto exclusivo de síntoma2.
Espectro de la MPS I
MPS I Grave (Hurler)

Mavish Hong Kong. Edad de los primeros síntomas: 6 meses. Diagnóstico de MPS I cuando tenía 1 año.
MPS I Atenuada (Hurler-Scheie)

Anisa, Canadá. Edad de los primeros síntomas: 2 años. Diagnóstico de MPS I a los 4 años.
MPS I Atenuada (Scheie)

Nick, EEUU. Edad de los primeros síntomas: 5 años. Diagnosticado de MPS I a los 9 años.
|
Características |
Hurler |
Hurler-Scheie |
Scheie |
|
Intensidad |
Grave |
Atenuada |
Atenuada |
|
Edad en el momento del diagnóstico |
De 0,2 a 7 años |
De 0,2 a 36 años |
De 2 a 54 años |
|
Efecto sobre la cognición |
Retraso mental pronunciado con pérdida de habilidades adquiridas |
Retraso mental leve o nulo; dificultad de aprendizaje |
Sin deterioro |
|
Esperanza de vida: media, mediana (sin tratamiento) |
6,8 años; 8,7 años |
Aproximadamente 20 años; NC |
Edad adulta; NC |
Referencias de la tabla:
Fallet S et al. JIEMS. 2014;2:5.
Moore D et al. Orphanet J Rare Dis. 2008;3:24.
MPS I Registry Boards of Advisors. Cambridge, MA: Genzyme Corporation; 2010.
Signos y síntomas de la MPS I
-
Hernias recurrentes (umbilicales/inguinales)
-
Cifosis/deformidad en giba
-
Tosquedad facial
-
Opacidad corneal
-
Hepatoesplenomegalia
-
Deterioro cognitivo
-
Trastornos del sueño/ronquidos
-
Alteraciones de las válvulas cardíacas
-
Baja estatura
-
Hernias recurrentes (umbilicales/inguinales)
-
Tosquedad facial
-
Contracturas articulares
-
Opacidad corneal
-
Trastornos del sueño/ronquidos
-
Hepatomegalia
-
Alteraciones de las válvulas cardíacas
-
Síndrome del túnel carpiano
-
Baja estatura
Los niños afectados por la MPS I grave (Hurler) presentan anomalías físicas y cognitivas evidentes poco después de nacer y experimentan un rápido deterioro, de forma que la mayoría de los pacientes no tratados fallecen en los diez primeros años de vida. Presentan un retraso del desarrollo y un deterioro cognitivo importantes, junto con una tosquedad facial característica, deformidades óseas y enfermedades respiratorias, cardíacas y hepáticas. 4
Signos más frecuentes*4,11
*No todos los signos y síntomas se observan en todos los pacientes
[lista tomada de Beck 2014, figura 2, síntomas observados en más del 50% de los pacientes]
Los niños y adultos con formas atenuadas de MPS I (Hurler-Scheie y Scheie) presentan anomalías físicas menos evidentes y un deterioro cognitivo leve o nulo. Las manifestaciones clínicas avanzan más despacio que en los pacientes gravemente afectados. En general, en los diez primeros años de vida aparecen síntomas como hernias, contracturas articulares o infecciones respiratorias. La aparición de estos síntomas, al ser característicos de enfermedades más frecuentes, provoca retrasos de varios años en el diagnóstico. 3-10 Los pacientes que presenten contracturas articulares en ausencia de inflamación deben someterse a una prueba de MPS I cuanto antes.1,9,10
Signos más frecuentes*4,11
*No todos los signos y síntomas se observan en todos los pacientes
[lista tomada de Beck 2014, figura 2, síntomas observados en más del 50% de los pacientes con Hurler-Scheie/Scheie combinados]
La MPS I, en cualquiera de sus formas, puede provocar la muerte prematura del paciente4


La enfermedad provoca trastornos importantes y, en algunos casos, muerte prematura4
Referencias: Guffon N, et al. Eur J Pediatr. 2019;178(4):593-603. Pastores GM, et al. Mol Genet Metab. 2007;91(1):37- 47. D’Aco K, et al. Eur J Pediatr. 2012;171(6):911-919.
Los gráficos que se presentan a continuación se basan en los datos recogidos en el registro de MPS I sobre la evolución natural de la enfermedad (Beck et al., 2014). En este análisis se incluyeron 955 pacientes, con una distribución global de fenotipos de 601 (60,9%) para el síndrome de Hurler, 227 (23,0%) para el de Hurler-Scheie y 127 (12,9%) para el de Scheie.

Figura 2. Prevalencia y edad de aparición de algunos signos y síntomas en pacientes con MPS I, por fenotipo. (a) Hurler, (b) Hurler-Sheie y (c) Sheie. Los porcentajes de frecuencia de los síntomas se muestran en el eje de la izquierda. Sólo se muestran los síntomas notificados durante el período de evolución natural en al menos el 25% de los pacientes con el fenotipo correspondiente. Los datos de edad equivalen a medianas de la edad en años (eje de la derecha) en los pacientes en que se registró la fecha de inicio de los síntomas. MPS I, MPS de tipo I.
- Hernia
- Rasgos faciales toscos
- Cifosis/giba
- Disostosis múltiple
- Opacidad corneal
- Hepatomegalia
- Trastornos del sueño/ronquidos
- Macroglosia
- Esplenomegalia
- Deterioro cognitivo
- Alteraciones de las válvulas cardíacas
- Hipertrofia amigdalina
- Contracturas articulares
- Hernia
- Rasgos faciales toscos
- Alteraciones cognitivas
- Macroglosia
- Trastornos del sueño / ronquidos
- Hipertrofia amigdalina
- Disostosis múltiple
- Contracturas articulares
- Opacidades corneales
- Hepatomegalia
- Cifosis/giba
- Esplenomegalia
- Alteraciones de las válvulas cardíacas
- Displasia de cadera
- Síndrome del túnel carpiano
- Edad de inicio (años)
- Hernia
- Contracturas articulares
- Disostosis múltiple
- Displasia de cadera
- Trastorno del sueño/ronquidos
- Rasgos faciales toscos
- Hepatomegalia
- Opacidad corneal
- Esplenomegalia
- Alteraciones de las válvulas cardíacas
- Síndrome del túnel carpiano
- Prevalencia
- Mediana de la edad de inicio
Todas las personas que aparecen en esta web han consentido la utilización de su imagen por parte de Sanofi.
MAT-ES-2301235 V1 Octubre 2023
1. Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, et al., eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill; 2001:3421-52.
2. Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 2009;123:19-29.
3. Vijay S, Wraith JE. Clinical presentation and follow-up of patients with the attenuated phenotype of mucopolysaccharidosis type I. Acta Paediatr 2005;94:872–77.
4. Beck M et al. Genet Med. 2014;16(10):759-765.
5. De Ru MH, Boelens JJ, Das AM, et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet J Rare Dis 2011;6:55
6. Clarke LA. The mucopolysaccharidoses: a success of molecular medicine. Expert Rev Mol Med. 2008;10:e1.
7. Arn P, Wraith J, Underhill L. Characterization of surgical procedures in patients with mucopolysaccharidosis type I: findings from the MPS I Registry. J Pediatr. 2009; 154:859-64 e3.
8. Moore D, Connock MJ, Wraith E, Lavery C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis 2008;3:24.
9. Cimaz R, Vijay S, Haase C, et al. Attenuated type I mucopolysaccharidosis in the differential diagnosis of juvenile idiopathic arthritis: a series of 13 patients with Scheie syndrome. Clin Exp Rheumatol 2006;24:196-202.
10. Thomas JA, Beck M, Clarke JT, Cox GF. Childhood onset of Scheie syndrome, the attenuated form of mucopolysaccharidosis I. J Inherit Metab Dis 2010;33:421-7.
11. De Ru et al. Orphanet Journal of Rare Diseases 2012, 7:22.
Temas relacionados:




La información contenida en este sitio web está dirigida exclusivamente a profesional sanitario facultado para prescribir o dispensar medicamentos en España (requiere una formación especializada para su correcta interpretación).
Sanofi promueve la prescripción de sus productos farmacéuticos en las condiciones establecidas en su ficha técnica.
Pulse ACEPTAR si usted es profesional sanitario en España y desea continuar en este sitio o SALIR para ser redirigido al sitio web de Sanofi.
campus.sanofi.es dice:
Hola, queremos avisarte que estás a punto de abandonar una web de Sanofi y accederás a otra página
web donde no se aplica nuestra política de privacidad. Sanofi no se hace responsable del contenido de dicha web externa.